Researchers have identified a new mode of action for Alzheimer’s disease (AD) using a new technique that measures the temperature inside individual cells called intracellular thermometry. The newly discovered cell pathology involves a protein called amyloid-beta clumping together to form toxic aggregates that overheat the cells’ interior—literally frying them.
How AD starts
The current thinking is that Alzheimer’s disease begins when amyloid-beta and tau proteins malfunction to form aberrant versions of themselves–starting as thin fibrils. These fibrils then spread their tendrils throughout the brain to clump together to form amyloid plaques and bundles of fibers known as neurofibrillary or tau tangles, respectively.
Collectively these toxic aggregates cause brain cells to die and the brain to shrink, resulting in memory loss, personality changes, and difficulty carrying out daily functions. Ultimately, the disease leads to complications from severe loss of brain function-such as dehydration, malnutrition, or infection-resulting in death.
Presently, the reason why these brain proteins begin to act erratically and attack the brain is still unclear. But, scientists suspect that a combination of genetic, lifestyle, or environmental factors may affect the brain over time to cause this neurodegenerative disease.
However, this is not set in stone as changes in the brain may begin a decade or more before symptoms appear, making it a difficult disease to study. A problem further compounded by the fact that clinicians can only give a definitive diagnosis after examining brain tissue samples after death.
New AD markers
Patients and their medical teams can sometimes miss the causes and progression of the disease. So you can imagine just how significant any new pathology would be to the research and development of possible treatments or cures for AD.
In an attempt to add to this body of work, a team from Cambridge University began investigating recorded instances of cancerous cells and lesions overheating. They did this as some forms of metastasis are similar to the initial spread of Alzheimer’s. So, the researchers applied these malignant examples to their AD model.
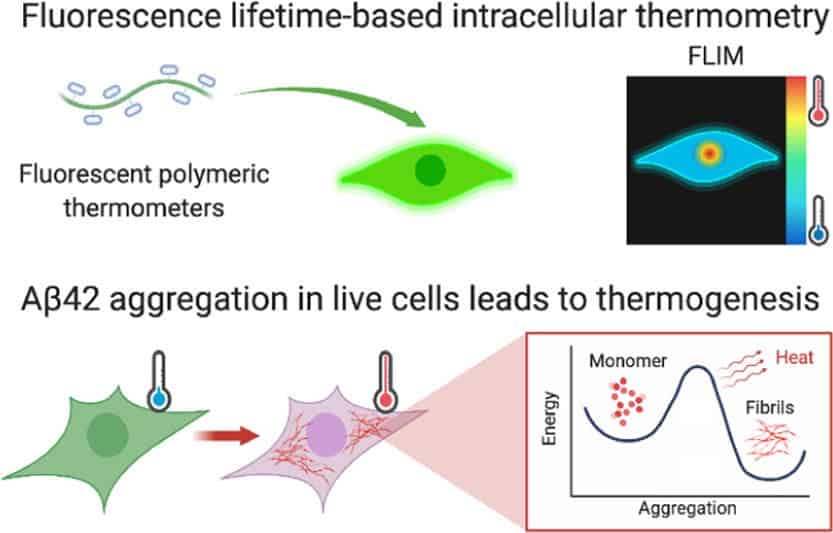
In the new study, scientists used miniature thermometers that were sensitive enough to measure the interior of live human cells cultured outside of the body in the laboratory. In their paper, the team says they witnessed the well-known Alzheimer’s protein amyloid-beta aggregate, causing the temperature to rise in the human cells in a process known as thermogenesis.
As a result, this process causes more of the healthy protein to aggregate, damaging the surrounding neurons and their connections. To counteract this, the group successfully employed a compound (MJ040) to lower the cell’s temperature and stop the amyloid-beta from aggregating.
“Thermogenesis has been associated with cellular stress, which may promote further aggregation,” said Chyi Wei Chung, the study’s first author. “We believe that when there’s an imbalance in cells, like when the amyloid-beta concentration is slightly too high and it starts to accumulate, cellular temperatures increase.”
“Overheating a cell is like frying an egg – as it heats up, the proteins start to clump together and become non-functional,” said Kaminski Schierle, who led the research.
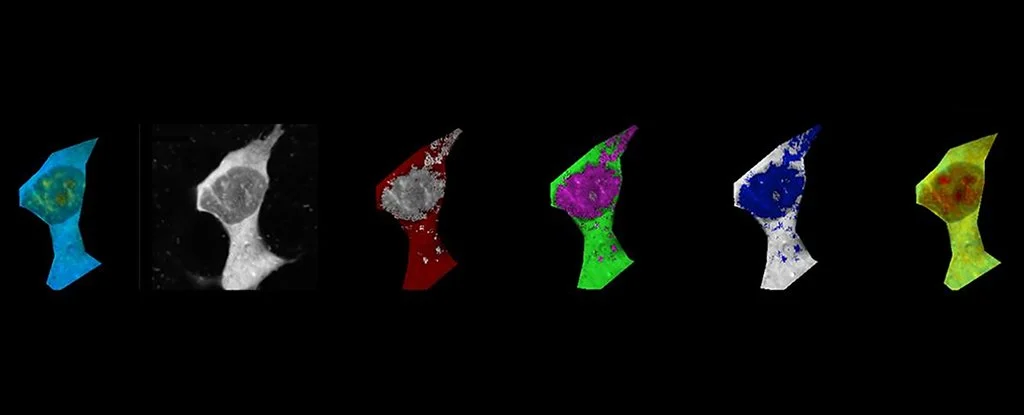
Tiny thermometers
To investigate the link between AD and cell thermogenesis, the team first added amyloid-beta to live human cell lines to begin the aggregation process. As the aberrant proteins formed, the researchers used tiny temperature sensors called fluorescent polymeric thermometers (FTPs) to measure the intracellular temperature over time. The FTPs worked by changing color to the corresponding temperature to provide a highly sensitive read-out for each cell.
FTP data showed that as amyloid-beta initially started to form thin fibrils, the average temperature of the cells began to rise. Indeed, measurements showed that the increase in cellular temperature was significant compared to cells with no amyloid-beta added.
“As the fibrils start elongating, they release energy in the form of heat,” said Kaminski Schierle. “Amyloid-beta aggregation requires quite a lot of energy to get going, but once the aggregation process starts, it speeds up and releases more heat, allowing more aggregates to form.”
“Once the aggregates have formed, they can exit the cell and be taken up by neighboring cells, infecting healthy amyloid-beta in those cells,” said Chung. “No one has shown this link between temperature and aggregation in live cells before.”
New AD drug
As well as inhibiting amyloid-beta aggregation in its tracks and lowering the temperature in cells to mitigate any further damage, MJ040 also helped the researchers to confirm that fibrils cause thermogenesis. This fact is important as scientists had always assumed that structures supplying cells with energy called mitochondria were causing cell thermogenesis in certain diseases. Accordingly, these data created much excitement, laying a longstanding mystery to rest.
However, because MJ040 inhibits the formation of amyloid-beta fibrils, the drug must be administered in the late stages of the disease. As can be imagined, this could cause problems if the patient is asymptomatic or in the advanced stages of AD. The team is also quick to stress that although their results suggest the compound has potential as a treatment for AD, extensive tests and clinical trials are required. Still, they surmise that overall the findings highlight MJ040’s potential as a therapeutic for AD.
But for now, their priority is to shed more light on the workings behind heat production caused by amyloid-beta aggregation. They hope their work could one day produce a diagnostic test for neurodegenerative diseases.
The team concludes: “We hope to spark off further work in this field to develop more accurate descriptors of complex intracellular phenomena that give rise to increased temperatures within a biological cell, which can be indicative of neurodegeneration.”
The study is published in the Journal of the American Chemical Society.
Was this helpful?


