Once upon a time, the idea that creatures too small to see made you sick was a concept that average people and doctors alike scoffed at. Think about it—if you didn’t already know about bacteria, wouldn’t the idea seem ridiculous? Without evidence, a person may as well have been telling you that tiny elves were attacking you. In relatively modern times, though, we have discovered that small organisms of all kinds are hard at work ruining our days. But as it turns out, whenever science thinks it has things figured out, something always steps up to the task of confusing us all. This time it was protein.
The Mystery
Now, we hear about bacteria and viruses all the time. More often than not, these are the causes of the illnesses we suffer. However, bizarre animal behavior, first officially noted in the 1730s, baffled scientists all the way through to the 1980s—and in the process of discovery turned our understanding of pathogens, and biology itself, on its head.
First, the Spanish observed strange behavior in their Merino sheep. The animals were walking strangely and pathologically scratching their bodies against fences. This led to the illness being called ‘scrapie’. Nearly two centuries later, in 1920, a disorder was noted in humans which caused degeneration of their brain tissue. This was named Creutzfeldt-Jakob’s disease, after the scientists who first noted this inexplicable phenomenon.
At the time, no one had noticed how similar the two diseases were. For one, they both had long incubation periods. In fact, in 1954 Sigurdsson proposed that scrapie was caused by a ‘slow virus’. No other form of pathogen known at the time fit this long incubation period. Scrapie took years to show symptoms, much like HIV. Meanwhile, in remote Papua New Guinea, an illness called kuru by the Fore people was officially noted—it looked a lot like Creutzfeldt-Jakob’s.
The Problem
By the 1960s, some scientists had begun to suspect that the agent of scrapie was not a virus at all but actually a protein. However, there was a very big problem—this directly went against everything we understood about cells, the central dogma of molecular biology. In simple terms, it states that information moves from DNA to RNA to form proteins and doesn’t go in the opposite direction. Essentially, proteins can’t give instructions, so how could one be a pathogen?
However, after many failed experiments designed to inactivate the illness—conducted by such scientists as T. Alper, J. S. Griffith, and I. H. Patterson—a protein was one of the only options left. Then, in 1967, Griffith officially proposed that scrapie was caused by a protein and even suggested possible mechanisms. Finally, in 1982 Stanley Prusiner coined the term ‘prion’, stating that the illness was caused exclusively by proteins. The speView Postcific protein involved was referred to as the PrP (Prion Protein). Prusiner’s research went on to earn him a Nobel Prize in 1997.
The Bizarre Reality
At last, we knew what was causing scrapie and eventually realized that kuru and Creutzfeldt-Jakob’s (CJD) were the same sort of illnesses. It was all down to prions, proteins which are able to change their shape and structure such that they can fold into non-functional shapes. All that was left to do was figure out just how a protein could cause disease.
Such diseases are called transmissible spongiform encephalopathies (TSEs) and, in these diseases, the brain degenerates in such a way that gaps begin to form in the tissue. In other words, they cause your brain to essentially become Swiss cheese or, as the name suggests, like a sponge in appearance. The results of this degeneration are severe and, unfortunately, always fatal.
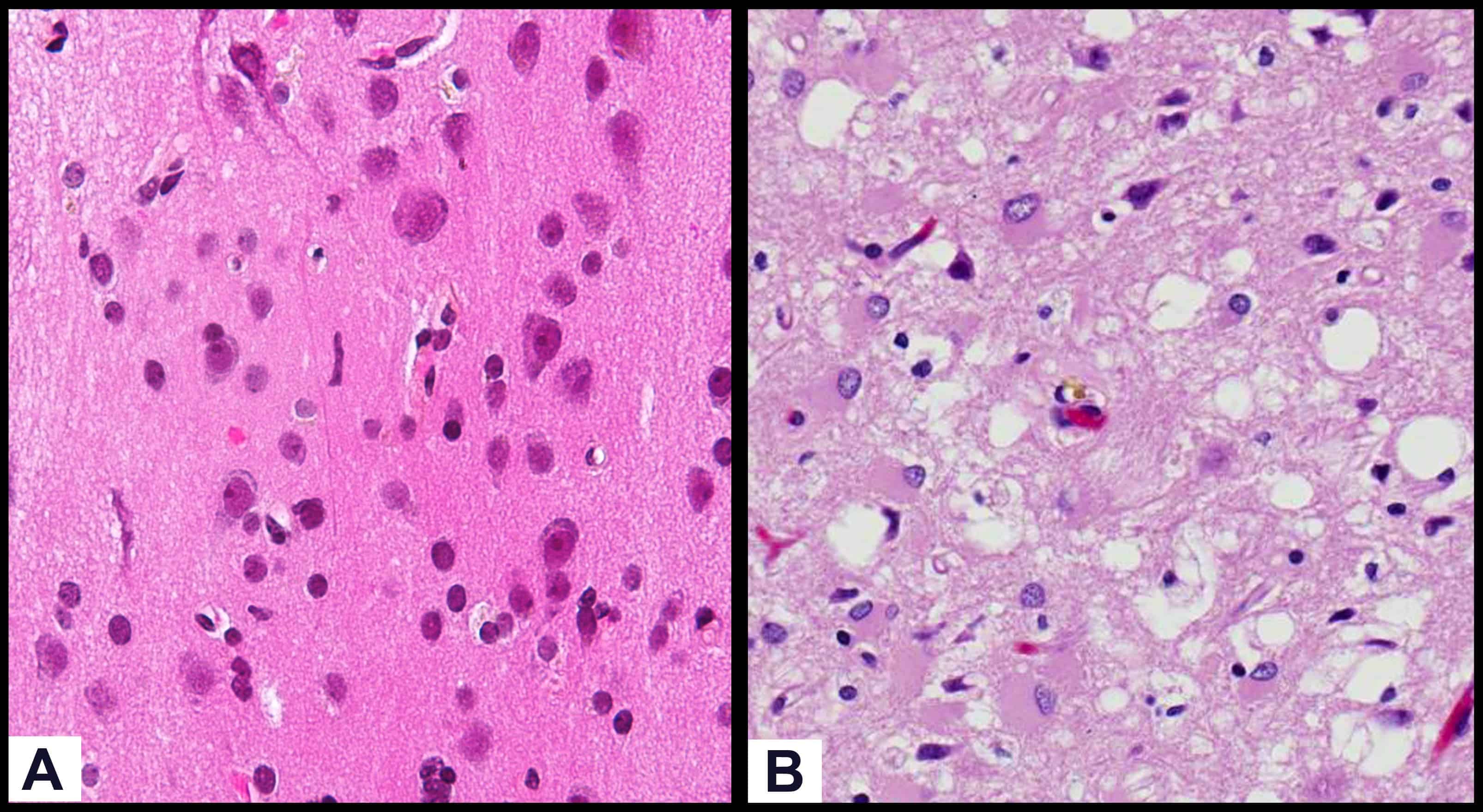
So how does a misfolded protein do all of that damage? The truth is that scientists still don’t really know the mechanism. However, a simplified way to look at it is that prions are highly charismatic proteins, able to ‘convince’ other proteins that their flawed structure is the right structure. The protein associated with most TSEs is called PrPC (for common), which is common in cell membranes in the normal configuration. It becomes a prion when it changes to PrPSC (for scrapie) which is the misfolded, infectious form of the protein.
Once consumed the PrPSC passes through the wall of the intestine by an unclear mechanism. Even more impressively, from there it manages to enter the central nervous system, apparently moving from nerve to nerve until it enters the brain itself. Upon encountering PrPC on a cell surface, it induces structural changes to occur, encouraging folding of the protein until it resembles the PrPSC form. Now satisfied that its work is done, the prion moves on to the next PrPC, showing itself off and spreading the good word about its bad conformation.

Before long it has gained a crowd following that is also going forth to share this new shape with others. The newly converted PrPSC tend to like to group together, forming aggregate communities throughout the brain. These aggregates form amyloid fibrils, insoluble clusters of protein which accumulate to form plaques (a process also associated with Alzheimer’s). And thus the spongiform structure occurs, with gaps being left behind by this accumulation. Worse still, this form of the protein is resistant to proteases, enzymes which break down proteins.
Some known prion diseases include kuru, scrapie, and CJD, as well as bovine spongiform encephalopathy (mad cow disease), and fatal familial insomnia (FFI). In most cases, the disease develops spontaneously with no apparent explanation. Otherwise, a person could consume tainted meat, such as in human cases of mad cow disease and kuru. In some forms, such as FFI, genes coding for the faulty protein can be further passed to offspring.
Symptoms of these illnesses include poor coordination, confusion, memory loss, hallucinations. In some cases, patients will experience dementia, psychoses and persistent insomnia. There are few ways to actively avoid such incredibly rare diseases but the best you can do is ensure that your food comes from safe sources—also, try not to eat any suspicious brains.
If you want to learn more and in more detail, you can take a look at the links below:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4626585/
- http://www.prion.ucl.ac.uk/clinic-services/information/prion-disease/
- https://www.cdc.gov/prions/index.html
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2805067/
- http://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.0020321
Was this helpful?